中国科学技术大学钱逸泰院士团队和王功名教授课题组通过实验和理论结合的方式,研究了金属钴基化合物(Co3O4, CoS2, Co4N以及CoP)在Li-S化学中的动力学行为,发现钴基化合物中阴离子的价电子的p能带中心相对费米能级的位置是影响Li-S电池界面电子转移反应动力学性质的主要因素。近日,该研究成果以《Deciphering the Modulation Essence of pBands in Co-based Compounds on Li-S Chemistry》为题,发表在Cell Press旗下的国际顶级能源材料期刊Joule杂志上。该论文第一作者是博士后周建斌,副研究员刘晓静和硕士研究生祝琳嵚。
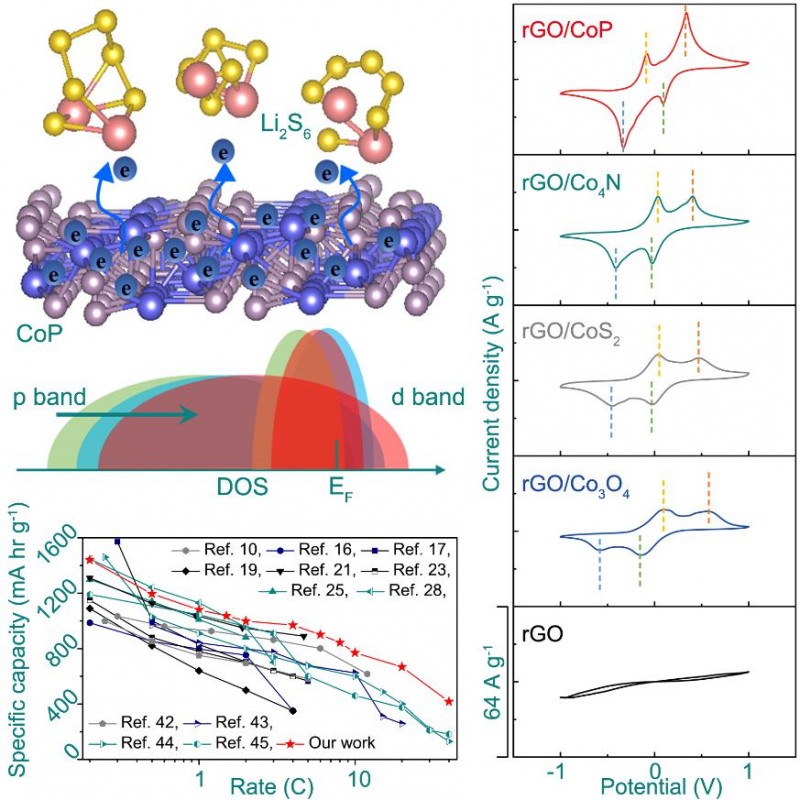
DFT模拟结果表明,相比于Co3O4, CoS2和Co4N,CoP对Li2S6以及Li2S的吸附能相对适中。同时电荷差分密度分析表明,吸附能与材料表面电荷的局域程度有很大的相关性;结合其电化学行为,发现过强或过弱的吸附能都不利于多硫化合物电化学转化的动力学性能,这与催化反应中的Sabatier理论一致;通过尝试关联不同钴基化合物的阴离子价带的p能带中心位置与多硫化合物电化学转化的动力学性能,发现改变阴离子价电子的p能带中心相对费米能级的位置,能够有效调控界面电子转移反应动力学,从而成为影响Li-S化学动力学性能的主要因素。
该项研究得到国家重大科学研究计划、国家自然科学基金等项目的资助。


